By Polly Clayden
NOTE: a postscript section at the end of this article includes periodic updates since publication in June 2012.
In the past year—since our last Pipeline Report—there has been a flurry of activity in pediatric antiretroviral drug development and approval.
While overall the pipeline for children looks encouraging, the short- and medium-term requirements of the youngest children in resource-limited settings still badly need to be addressed. What is currently available to treat them has been described as, “too many formulations and yet too few real options.”[1] At present, national programs use over 45 single agents and co-formulations due to an initial requirement by many programs to use single innovator formulations. Meanwhile the market is very small, and further fragmented by different regimens across age groups and weight-band doses. As a result, orders are always low-volume, and this threatens both access and the sustainability of the market. Therefore, the focus needs to be on a smaller number of products that offer the best options for children.
Several of the pipeline compounds might offer advantages over currently available options, and work is ongoing to make formulations of already approved drugs that are more suitable. For children less than three years old particularly, promising public-private partnerships may help to deliver appropriate drugs and regimens.
New Approvals
Over the past few months, we have seen the following approvals of new drugs from innovator companies by the United States Food and Drug Administration (FDA):
- On December 16, 2011, a 100 mg/mL oral suspension formulation of darunavir, and dosing recommendations for children 3 to less than 6 years of age (and children 6 and older who are unable to swallow darunavir tablets).[2] There is a waiver for children under 3, due to very high darunavir concentrations in animals (of an analogous age) and, in turn, toxicities in preclinical studies.
- On December 21, 2011, a 100 mg scored chewable tablet and a 25 mg chewable tablet of raltegravir, and dosing recommendations for children 2 to 18 years of age and weighing at least 10 kg.[3] Studies with a new granule formulation for very young children are under way.
- On January 18, 2012, an oral powder (40 mg per 1 gram of oral powder) formulation, and 150 mg, 200 mg, and 250 mg tablets of tenofovir, anddosing recommendations for children 2 to less than 18 years of age.[4]
- On March 26, 2012, a scored 25 mg etravirine tablet, and dosing recommendations for treatment-experienced children 6 to 18 years of age and weighing at least 16 kg.[5] Studies in the younger age groups are planned. Etravirine might also be a useful second-line non-nucleoside reverse transcriptase inhibitor (NNRTI) option for children as its resistance profile is different from those of nevirapine and efavirenz; it should not, however, be co-administered with rifampicin.
- On April 27, 2012, an oral suspension of fosamprenavir was approved for use in children 4 weeks to less than 6 years of age.[6] It is not expected to be used widely in young children.
These approvals by the FDA are welcome, and kick off the painstaking process that will eventually lead to access for children in the regions that need them the most. The execution of this will require commitment from many entities including the World Health Organization (WHO); national departments of health; local regulatory agencies; innovator and generic manufacturers; UNITAID and other donors; and non-governmental agencies (NGOs) such as the Clinton Health Access Initiative (CHAI), Drugs for Neglected Diseases initiative (DNDi), and Médecins Sans Frontières(MSF).
Darunavir
Boosted darunavir is generally considered to be the most durable protease inhibitor (PI) for adults. It is increasingly used in children and adolescents in industrialized countries, particularly in those with treatment experience.[7] Boosted darunavir could be a useful option for third-line regimens for children, and for second-line regimens where boosted lopinavir has been used as first-line.
The Pediatric Antiretroviral Group of the WHO considers darunavir to be of high priority, and the 2011 Updated List of Missing Drug Formulations lists a tablet or sprinkle formulation of darunavir/ritonavir as urgently needed.[8]
However, using boosted darunavir with currently approved doses does not lend itself to harmonized, simplified weight-band dosing or to appropriate use in combined tablets to facilitate this. The establishment of a single ratio at best, or at least a simpler dosing range would make wider use of darunavir more feasible. As the varied ratios were because of the limits of ritonavir formulations, there seems no reason why a 6:1 ratio twice daily, as for adults, shouldn’t be possible. See Table 1.
TABLE 1. Darunavir/Ritonavir Dosing Recommendations for Children >3 Years Old
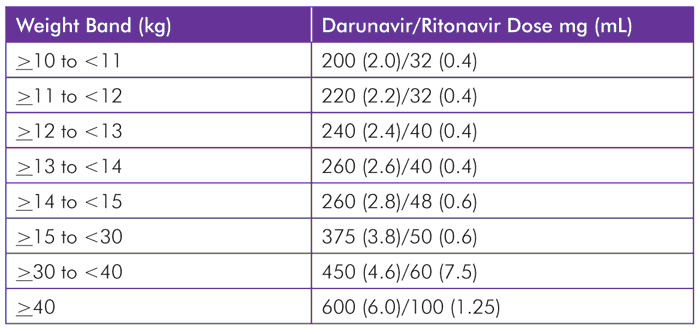
Children <15 kg: oral suspension, and >15 kg: reduced-strength tablets (150 mg and 75 mg)/oral suspension if they cannot swallow tablets
Raltegravir
Raltegravir’s approval ushers in a new therapeutic class—integrase inhibitors—for young children that might offer some advantages over the currently available drugs. Granule formulations are more desirable for resource-limited settings as they are easier to use, transport, and store than suspensions, but still not nearly as convenient as scored, reduced-strength tablets. Alongside darunavir, raltegravir has also been suggested as a future option for third-line treatment and a high priority for children. But alongside darunavir, it is currently very expensive, with no generic options yet—even for adults.
Tenofovir
Tenofovir approval for children has been a long time coming; the FDA approved it for adults in 2001. Tenofovir is currently the preferred nucleoside reverse transcriptase inhibitor (NRTI)/nucleotide reverse transcriptase inhibitor (NtRTI) for adults in U.S., European and WHO guidelines. Both problems with a suitable formulation and concerns about toxicities have delayed its pediatric approval.
The original liquid-suspension formulation developed for children was too bitter-tasting for further development. Although the newly approved powder for younger children is an improvement, its unpleasant taste is not well masked, and it is difficult to administer and hinders adherence. Reduced-strength tablets appear to be more palatable, although exposure can be variable with the approved dose.[9]
The innovator company Gilead has experience working with generic manufacturers to produce lower-cost tenofovir, including as part of fixed-dose combinations (FDCs). It may be possible to produce pediatric FDCs or suitable formulations of scored adult tablets of tenofovir/3TC/efavirenz so that first-line treatment of children over 3 years old could be aligned with that of adults. Tenofovir given in this combination achieved exposure in children 3 years and older, dosed according to WHO weight-band tables, comparable to that in adults.[10]
However, remaining uncertainties over tenofovir-associated decreased bone mineral density have been a regulatory hurdle. There have been concerns about this in growing children and adolescents. Renal tubule dysfunction and glomerular toxicity do not appear to differ between children and adults.
Although the FDA approved it, the European Medicines Agency (EMA) initially rejected tenofovir for the 12-to-18-year age group due to concerns about poor efficacy (shown in the studies presented) and concerns about long-term bone toxicity. The agency agreed to a pediatric investigation plan both for the single agent and the investigational tenofovir-containing FDC (subject to more information on individual drugs). The decision from this agency is still pending.[11].[12]
Tenofovir has long been considered a high priority for children in resource-limited settings, particularly with respect to harmonization with adult treatment. In addition, the cost of abacavir is too high, zidovudine is associated with anemia and, not least, stavudine-related toxicity in children appears similar to that seen in adults.[13],[14]
The WHO has recently published a technical update on use of tenofovir in children,[15] including review of published manuscripts and unpublished data from the innovator company. If the WHO recommends its use in children over two, it would be possible for programs to align first-line treatment from three years old to adulthood.
Targeting the Youngest Children
Less than half of FDA-approved adult antiretrovirals are approved for neonates and infants. See Table 2.
WHO and national guidelines recommend universal treatment for infants and children up to two years old.[16] Conventional drug development does not always serve this population well, even with considerable incentives from the regulatory agencies.
TABLE 2. Pediatric FDA Antiretroviral Approvals by Age Group (Years)
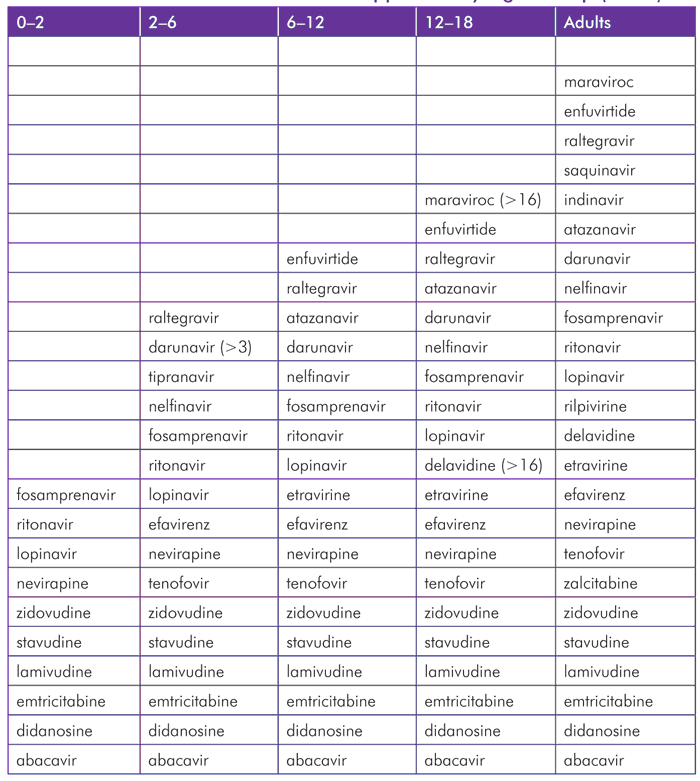
New agents are studied in children in de-escalating age bands (but this needs to be challenged), and appropriate formulations are not always easy to develop—it can require formulating often quite big and/or insoluble molecules into appropriate forms—so even if an indication for those less than two years old is eventually approved, the process can take several years and, strictly speaking, the indication for several antiretrovirals is not from birth, but 2 to 4 weeks (and abacavir is from 12 weeks).
Traditionally, liquid formulations were developed for this age group. These are expensive, have short shelf lives, and are hard to store and transport.[17] Some of the early pediatric programs in resource-limited settings used divided adult FDCs to treat children, which can be effective in older children but can lead to inexact dosing in younger ones, who also require different ratios of some drugs in FDCs.[18]
First FDCs for Children
In order to address this, Cipla developed reduced-strength, dispersible, scored FDC tablets of nevirapine/stavudine/lamivudine (Triomune Baby and Junior) with ratios of drugs appropriate for young children and doses appropriate for WHO weight-band dosing, which were FDA-approved and accepted for WHO prequalification in 2008.[19] These formulations made it possible for programs to begin treating children in places where liquids posed too many problems.
For infants who have been exposed to NNRTIs to prevent vertical transmission, WHO and national guidelines recommend a protease inhibitor. Data from The IMPAACT P1060 trial—which showed about 20% higher rates of failure in children ages two months to three years who received NNRTI-based regimens compared to PI-based, whether or not they had been NNRTI-exposed—suggest that this recommendation might be made for all children less than two years in the next guideline revisions.[20],[21] There is some concern with these results though as the trial followed children for only 24 weeks, and lopinavir/ritonavir is unpalatable in its current formulation. Nevirapine is currently more widely used in children less than two in resource-limited settings.
New Formulation of Lopinavir/Ritonavir
Cipla is working with the Children with HIV in Africa – Pharmacokinetics and Adherence of Simple Antiretroviral Regimens (CHAPAS) group at the Medical Research Council Clinical Trials Unit, the Joint Clinical Research Centre, and Baylor International Pediatric AIDS Initiative (BIPAI) Uganda to produce a more acceptable new formulation of lopinavir/ritonavir. The initial results of the CHAPAS-2 trial on pharmacokinetics and acceptability of sprinkles in babies less than one year old and children aged four and above, funded by the Monument Trust in the United Kingdom, show comparable exposure to lopinavir/ritonavir from sprinkles to syrup in infants; it was lower than tablets in older children. The caregivers found the sprinkles were more acceptable for infants but not for older children, mainly due to the taste.[22]
Drugs for Neglected Diseases Initiative (DNDi)
Last year, DNDi entered the pediatric HIV arena.[23],[24] DNDi is a not-for-profit research and development organization that develops new drugs for neglected diseases such as human African trypanosomiasis, visceral leishmaniasis, Chagas disease, and malaria. DNDi was asked by various organizations, including MSF and UNITAID, to turn its expertise to pediatric antiretrovirals for children less than three years old. In consultation with experts, DNDi developed “ideal” and “acceptable” specifications for desired formulations or regimens (see Table 3) alongside priorities for appropriate research to facilitate their approval.
TABLE 3. Target Product Profile (TPP) for Children Less than 3 Years Old
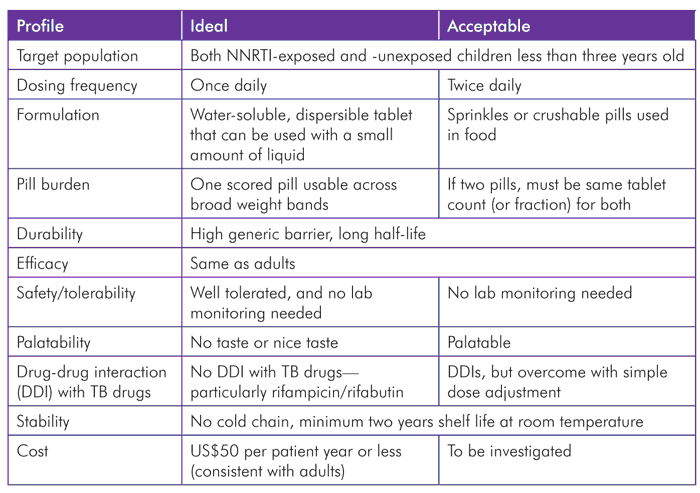
DNDi is currently working on the development of a regimen of granule formulations in sachets using ritonavir-boosted lopinavir and two NRTIs. For infants receiving concomitant treatment for tuberculosis, an additional dose of ritonavir can be added; this is to overcome the drug-drug interaction between rifampicin and lopinavir/ritonavir—rifampicin dramatically reduces plasma lopinavir/ritonavir concentrations coformulated in a 4:1 ratio.[25]
They are also supporting the one-to-four-year-old cohort of CHAPAS-2.
Their plan is to have the new regimen by 2015 and to consolidate rather than further fragment the market—that is, to have this regimen replace some of the many existing formulations currently available for infants and young children.
An Innovator Company Enters the (It Would Be Nice to Say) Fray
A recent announcement of a novel public-private partnership of ViiV Healthcare, CHAI, and the Indian generic company Mylan Inc. also offers a new model that could deliver a suitable product for young children.[26]
For this initiative, CHAI has identified the ideal characteristics of a pediatric formulation of two backbone NRTIs. ViiV will oversee and fund the development of a dispersible tablet formulation of abacavir/lamivudine. Then the company will transfer the technology and resources to Mylan for production, registration, and distribution at the lowest possible cost for low-income, least-developed countries.
The tablet will be dosed according to weight, thus it will be one tablet, two tablets, three tablets, or four tablets, with four being the highest for the largest body-weight required before an infant can use an oral tablet. The dosing is being confirmed through bioequivalence studies.
Although there are already combination products with these two drugs for children, dispersible tablets are most useful.
This new formulation tastes of strawberries and is suitable for children from 12 weeks old.
Filing with the FDA is planned for July 2013 and approval expected in early 2014.
THE PIPELINE
Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
Etravirine
The recommended dose per weight band for children and adolescents ages 6 to 17 is based on 5.2 mg/kg twice daily. The FDA recently approved dosing recommendations for etravirine for treatment-experienced pediatric patients 6 to 18 years of age and weighing at least 16 kg as well as the scored 25 mg tablet.[5]
IMPAACT P1090 will evaluate the drug in treatment-naive and -experienced children ages 2 months to 6 years.[27]
Rilpivirine
The PAINT trial is currently recruiting treatment-naive adolescents ages 12 to18, weighing more than 32 kg, and receiving 25 mg once daily plus two nucleoside reverse transcriptase inhibitors (NrTIs). The trial will evaluate steady-state pharmacokinetics and short-term antiviral activity in this age group.[28]
TMC278-C220 is an open-label single-arm trial using the granule formulation, planned in children ages 2 to 12 years. This trial is taking a staggered approach and will study the drug in de-escalated age groups, down to 2 years of age.
Protease Inhibitors (PIs)
Atazanavir
The capsule formulation is approved in the United States for children ages 6 years and older who are treatment-naive and weigh 15 kg or more, and for treatment-experienced children weighing 25 kg or more. In the European Union, it is approved for both treatment-naive and treatment-experienced children ages 6 years and older and weighing 15 kg or more.
Treatment-naive and -experienced children ages 3 months to 6 years receiving atazanavir unboosted and boosted with ritonavir are being studied in PRINCE 1 and 2 and IMPAACT P1020A.[29],[30],[31] PRINCE 1 is now fully enrolled, and data are expected in early 2013; PRINCE 2 is 50% enrolled, and data are expected at the end of 2013. Safety and PK data in this age group are urgently needed; the atazanavir/ritonavir ratio is 3:1 and as with darunavir this is complicated by the currently available formulations.
Lopinavir/Ritonavir
The generic manufacturer Cipla is developing a sprinkle formulation of lopinavir/ritonavir. The formulation (40/10 mg lopinavir/ritonavir) consists of a finite number of mini-tablets in a capsule, which is opened and sprinkled on soft food.
Data from a randomized crossover pharmacokinetic study in healthy adults comparing a single dose of sprinkles from 10 capsules of lopinavir/ritonavir with a single dose of 5 mL Kaletra oral solution (each mL containing 80 mg lopinavir and 20 mg ritonavir) were recently presented.[32] Both formulations were administered with about 150 g porridge and 240 mL water.
Most of the pharmacokinetic parameters fell within the conventional bioequivalence range of 80–125% in this study. Where they fell outside, the differences were not large.
Initial data from CHAPAS-2—which compared twice-daily sprinkles to tablets in children ages 4 to 13 years, and sprinkles with syrup in infants ages 3 to 12 months in a randomized cross-over PK study—found high variability in the younger cohort with both sprinkles and syrup, with no significant difference in subtherapeutic concentrations between formulations. In the older children, lopinavir/ritonavir concentrations were lower in children receiving the sprinkles than in those who got the tablets.[33]
Acceptability data showed storage, transport, and conspicuousness were less problematic for sprinkles compared with syrups, but for older children, several caregivers commented about the number of capsules needing to be used.
At week 8, when they could chose which formulation to continue with, 10 out of 14 (71%) caregivers chose to continue sprinkles rather than syrups for the infants, but only 7 of 29 (24%) of the older children chose sprinkles over tablet, and taste was particularly to blame.
The study comparing syrups to sprinkle in one- to four-year-olds is ongoing.
Integrase Inhibitors
Dolutegravir
The IMPAACT P1093 study will work with de-escalated age bands of children down to six-week-old infants. The older children will receive tablets, and the younger ones the pediatric formulation.
A granule formulation has been developed, and results from a phase I pharmacokinetic study in healthy adult volunteers were recently presented.[34] The granules were given with and without 30 mL of various liquids and compared to the current tablet formulation given with 240 mL of tap water.
Subjects received a single dose of dolutegravir as a 50 mg tablet (adult formulation) and as 10 g of granule given: direct to mouth with no liquid; with purified water; with mineral water containing high-caution concentrations; or with infant-formula milk.
Dolutegravir exposures of the granule formulation were all moderately higher than those of the tablet formulation, with or without liquids. Exposure was highest when the granule formulation was given with formula milk.
The granule formulation is being studied further in children in IMPAACT P1093.
A reduced-strength FDC of dolutegravir (DTG), abacavir (ABC), and lamivudine is also planned.
Elvitegravir
The 183-0152 study was a phase Ib open-label nonrandomized trial in treatment-experienced adolescents receiving 150 mg once daily plus a PI-optimised background regimen. Of the 21 subjects enrolled in the 10-day pharmacokinetic study, 9 of 11 eligible subjects continued elvitegravir plus a ritonavir-boosted PI-containing optimized background regimen, and completed 48 weeks of treatment.
The pediatric committee of the EMA granted a positive opinion to the cobicistat and Quad pediatric investigational plan in April 2011.
Boosted elvitegravir will be studied in de-escalated weight bands, and a suspension formulation is in development for the youngest children.
The Quad study will start after a review of data for elvitegravir and cobicistat. Age-appropriate formulations are planned.
Raltegravir
The adult 400 mg film-coated tablet is approved in the United States for use in adults and in children ages 6 to 18 weighing >10 kg, and 100 mg and 25 mg chewable tablets are approved for children >2 to <12 years old at a maximum dose of 300 mg.
The pediatric program is ongoing in IMPAACT P1066, and an oral granule formulation is being studied in the youngest children and babies down to 4 weeks old. Intensive PK and preliminary 24-week safety and efficacy data on 6-month- to <2-year-olds receiving the raltegravir oral granule formulation were recently presented.[35]
In this dose-finding study of treatment-experienced children, they received weight-based raltegravir oral granule suspension at ~6 mg/kg, twice daily.
The PK values achieved were similar to those observed in 2-year-old to <12-year-old children receiving chewable tablets; at week 12, 78% of the 9 children achieved virological suppression, and by 24 weeks, 85% achieved virological suppression.
The dose of 6 mg/kg every 12 hours was chosen for continued study in this age group.
Raltegravir also has the potential for use as prophylaxis to prevent vertical transmission to infants, and for treatment of HIV-infected infants. IMPAACT P1097 is an ongoing phase IV washout (passive) PK and safety study of infants born to women who received at least two weeks of raltegravir (400 mg twice daily) in pregnancy and through labor. This is the first clinical trial of an investigational antiretroviral to look at neonatal pharmacokinetics. Raltegravir crosses the placenta well. It is metabolized primarily by an enzyme in the liver (UGT-1A1) that is immature in neonates. UGT pathways increase in activity hugely in the first weeks of life.
Cord blood and single maternal blood samples were obtained within one hour of delivery alongside infant blood samples up to 36 hours after delivery. Early data from this study from nine mother-infant pairs showed maternal concentrations at delivery similar to those in cord blood (113%). The mean cord blood to maternal delivery concentration ratio was 1.14 (55%). The mean last infant raltegravir concentration at 30–36 hours was 407 ng/mL (range: 42.1–1,401 ng/mL). The mean neonatal half-life was 23.2 hours (range: 9.3–87.8 hours). No safety issues were identified at 20 weeks of life from in utero and transplacental exposure.[36]
Simulations combining these data plus PK data from four-week-old to six-month-old babies in P1066 will be used to determine the dose and frequency for neonates. The data from the washout study suggest that it may be possible to initially dose raltegravir once daily in newborns.
CCR5 Receptor Antagonist
Maraviroc
The A4001031 study is ongoing in children 2–18 years old who are infected with the CCR5-tropic virus (virus variants that use the CCR5 receptor for entry). Use of this drug requires an expensive tropism assay, as it will not work for people with the CXCR4-tropic virus or in dual- ormixed-virus (CCR5/CXCR4) populations.[37]
Preliminary data in 29 children showed body surface area–based doses of maraviroc provided adequate exposures when administered with a protease inhibitor as part of their background regimen. Children who were not receiving a boosting agent in their background regimen required at least doubling of the initial dose.[38]
A body surface area–scaled twice-daily tablet dose of maraviroc in treatment-experienced children 6 years and above concomitantly receiving boosted protease inhibitors (darunavir and lopinavir) achieved concentrations similar to those in adults receiving 150 mg maraviroc twice daily with a boosted protease inhibitor.[39]
TABLE 4. Pediatric ARV Pipeline
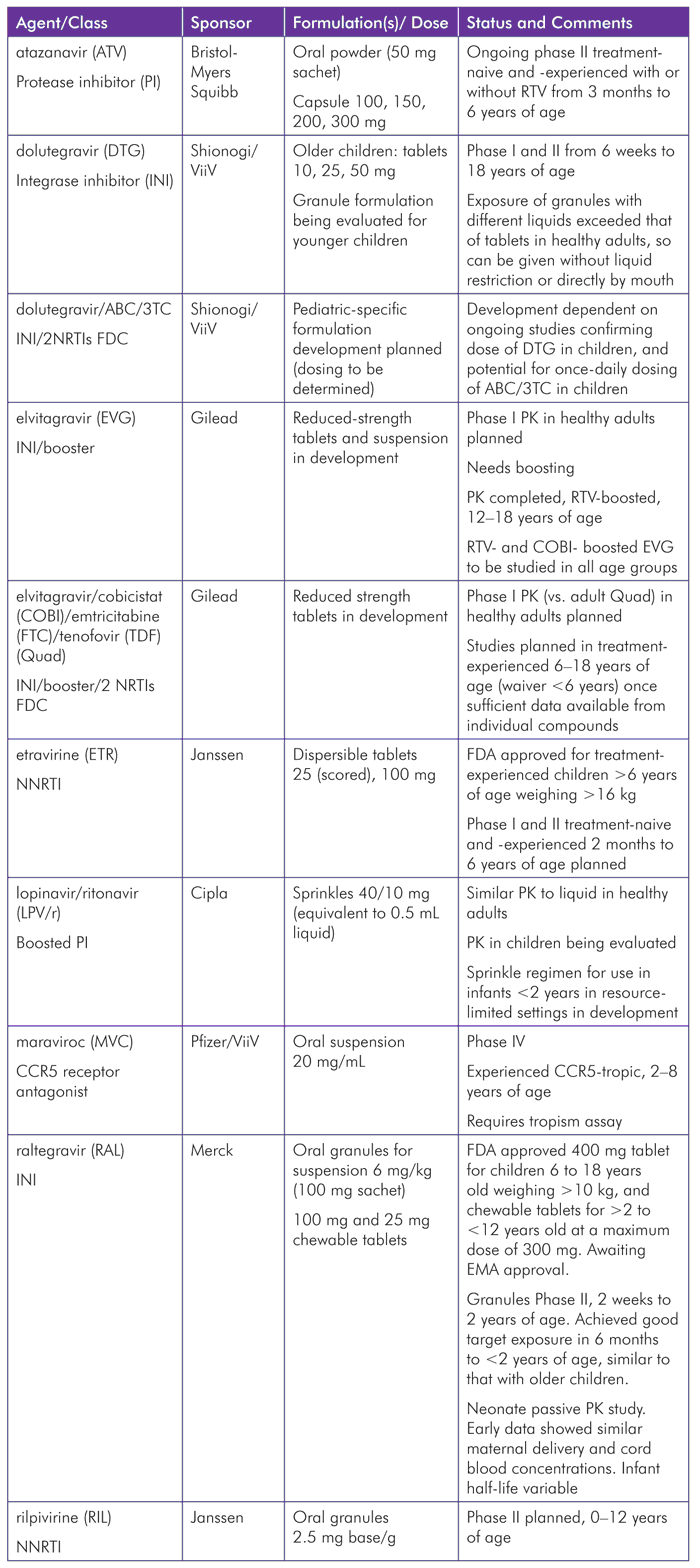
Discussion
Despite the ever-diminishing pediatric antiretroviral market in industrialized countries, with little return on investment in research and development to produce what are often complex formulations, the pipeline looks quite promising.
But, as shown with tenofovir, which was finally approved for children down to two years of age, 11 years after its approval for adults; atazanavir, for which studies in the youngest age group have been painfully slow to recruit; and efavirenz, which is complicated to use in children less than three years old (for whom it appears that its optimal use requires pre-treatment genotyping and genotype-guided dose optimization), there are clearly technical, physiological, and operational barriers to developing appropriate formulations for young children in a reasonable time frame.[40],[41]
On a more optimistic note, for some of the newer antiretrovirals, things do seem to be moving more swiftly than in the past. This might be a result of regulatory incentives and penalties: since 2007, EMA marketing authorization cannot be granted for a new medicinal product without a Pediatric Investigation Plan (PIP).[42] This obligation comes with rewards, like six months’ extension of patent protection. The PIP must be submitted when the adult phase I is completed and include details of methods proposed—with all pediatric subsets covered by a combination of studies and waivers— including age-relevant dosing and formulations. Although not so strict, the FDA has similar incentives.[43] Whether or not this has moved things along is a moot point, but regulations do seem likely more effective than reliance on what remains a vulnerable market or industry largesse.
Furthermore, formulations for young children for all but one drug in the current pipeline are granules, a dispersible tablet, or a powder, which might be useful for resource-limited settings in the future, although still not as desirable as dispersible scored tablets.
The work for children by the generic company Cipla is laudable and continues to be pioneering.
The work by DNDi is very important, as it specifically targets the youngest age group of children living in these settings.
The ViiV/CHAI/Mylan plans are also promising, not least as a rehearsal for future strategies. In a separate chapter of this report, Simon Collins describes some of the advantages of dolutegravir—low milligram dose, no boosting required, low pharmacokinetic variability, etc.—which will also apply to children’s formulations. A granule formulation is already in development, and an FDC is planned. Further along the pipeline, the follow-up integrase inhibitor S/GSK-1265744, under investigation as a long-acting formulation, has provoked interest as a potential treatment of adolescents (as has the long-acting formulation of rilpivirine). If these compounds do fulfill their early promise, the company should use its experience with the dual-nucleoside formulation, figure out the intellectual property issues, and transfer the technology and resources to a generic company for production, registration, and distribution at the lowest possible cost for low-income, least-developed countries. And other companies should take note.
What Needs to Be Done?
Reality check: although antiretroviral treatment coverage for adults continues to grow and now reaches about half those currently in need, for children—despite a dramatic increase in those receiving antiretrovirals since 2005—this proportion does not even reach a quarter. So the remaining majority of the 3.4 million children in need of treatment worldwide are still neglected.
Meanwhile pediatric HIV is becoming an old story set against a backdrop of targets to eliminate vertical transmission by 2015, which though they are to be applauded, must not happen at the cost of continual scale-up for children.
In order for this not to continue, a number of things urgently need to be addressed:
- Definitive guidance. The next WHO guideline revision is likely to recommend a lopinavir/ritonavir-based regimen first line irrespective of NNRTI exposure for all children under three. Older children might be able to align with adults and receive efavirenz/tenofovir/lamivudine. Guidance is also needed for second-line treatment.
- Research gaps. There is still uncertainty with regard to tenofovir use in children. The release of the WHO systematic review is important, as is analysis of existing cohort data. Whether NNRTI-exposed children can switch from lopinavir/ritonavir to an NNRTI following early treatment with a boosted protease inhibitor is unclear with a switch to nevirapine, but ongoing work in NEVEREST-III will shed light on whether or not this is possible with efavirenz.[44],[45]
- Suitable formulations.Development, approval, and distribution of new formulations need to happen in ways that are timely and do not further fragment the market. The time from first FDA/EMA approval to when products are available where they are most needed must shorten. This will require earlier access by generic companies to new products (which must include the possibility to develop FDCs with components from different innovators) and registration by the WHO and in country. To reduce the current situation with too many formulations and too few real options,products need to be rationalized and unsuitable ones phased out.
- Consolidated procurement.CHAI needs to continue with its successful model of price negotiations.[46] Concerted efforts by international donors, including the Global Fund and PEPFAR, need to be made to facilitate the transition from previous reliance on UNITAID funding of pediatric products. In the many individual countries where orders do not meet manufacturer volume requirements, buyers must get together.
Postscript updates
August 2012
- At IAS 2012, the first pediatric data for dolutegravir from the adolescent cohort (aged 12 to 18 years) of the IMPAACT P1093 study was presented. [47] As was the data to support the recent approvals for raltegravir, etravirine and fosamprenavir [48, 49, 50] Link to i-Base article http://i-base.info/htb/19859
- Data from CHAPAS 2, showing pharmacokinetics and acceptability of the Cipla generic lopinavir/ritonavir sprinkles was notable for its absence at the IAS conference, but presented at the preceding pediatric workshop. [51] DNDi will work with Cipla to develop a 4-in-1 pediatric regimen using this formulation alongside two sprinkle formulation NRTIs. [52] Link to i-base article http://i-base.info/htb/19902
References