The basics of generic medications, bioequivalence, and the push for good manufacturing practices
Tim Horn
Securing access to generic drugs to treat HIV, hepatitis C virus (HCV), and tuberculosis (TB) is now one of the most prominent strategies of global health care and treatment activism.
In the vast majority of low-income countries, the licensing of generic antiretrovirals (ARVs) is a key driver behind the 40-fold increase in treatment access for people living with HIV since 2002. In high-income countries, particularly the United States, a confluence of skyrocketing brand-name (originator) drug costs and the approaching expiration of patents protecting several commonly used ARVs has led to a tremendous interest in the potential cost savings and acceptability of HIV treatment regimens with generic components.
Effective responses to the entrenched TB epidemics are also dependent on affordable and consistent access to generic antimicrobial agents (see Kenyon Farrow’s “Safeguarding against Stock-Outs,” and Erica Lessem’s “Generics vs. the Giant”). Moreover, with the arrival of short-course, all-oral curative—but expensive—therapy for HCV, there is mounting interest in generic equivalents to new originator drugs to ensure that all those who need these lifesaving therapies, no matter where they are in the world, have affordable access to them (see Karyn Kaplan’s and Tracy Swan’s “The Road to Treatment Access”).
The ongoing development, regulatory approval, and evaluation of generic drugs are dependent on activism. This requires a basic understanding of the science and policies of generics, particularly the practices that must be followed to help ensure equivalence and quality control.
The World Health Organization defines a generic drug as a “pharmaceutical product, usually intended to be interchangeable with an [originator] product, that is manufactured without a license from the [originator] company and marketed after the expiry date of the patent or other exclusive rights.” This is mostly accurate, though generic versions of patent-protected originator ARVs have been produced through voluntary or compulsory licensing pathways (and in countries where international patents are not recognized, particularly for older HIV drugs), with similar approaches being eyed for HCV and TB drugs as well.
For many generic drugs, particularly oral and injectable medications that work systemically, establishing equivalence to innovator products is a fairly straightforward process. First and foremost, a generic drug must contain the same active pharmaceutical ingredient (API). It must involve the same route of administration (e.g., oral), formulation (e.g., capsule or tablet) and dosing. It must also meet stringent criteria for bioequivalence—the extent (and, often, the rate) of absorption must not differ significantly from that of the originator drug. A generic drug that meets these standards should not behave any differently, either in terms of efficacy or safety outcomes. (Medications that work topically or locally, such as ointments or ophthalmology drugs, and biologics that use active substances derived from living sources such as cells, including interferons and monoclonal antibodies, must meet other criteria to prove equivalence.)
Bioequivalence is assessed in studies, often involving 20 to 50 human volunteers without the infection for which the drug is indicated, and requires comparing a series of blood samples collected in the minutes, hours, and days after sequentially administering single doses of the originator and generic drugs (see figure).
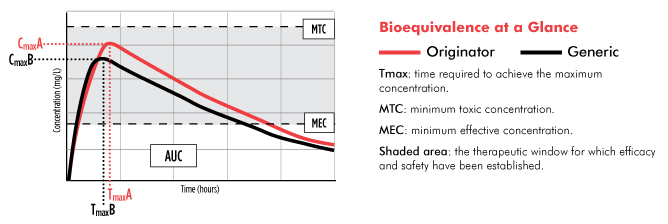
Of greatest interest to generics manufacturers and regulatory agencies, such as the FDA, are two measures of bioequivalence: the maximum concentration of the drug (Cmax) and the total extent of drug absorption (the area under the cure, or AUC).
To be considered bioequivalent, a generic drug’s Cmax and AUC do not need to exactly match that of the innovator drug. While some sources note that the FDA only requires the extent of a generic drug’s concentration (Cmax and AUC) to be within 80 to 125% of that established for the innovator drug—a difference of 45%—this is something of an oversimplification. More accurately, the 90% confidence intervals for the ratio of the Cmax and AUC mean averages must be in this range. In fact, according to a meta-analysis published in 2009, a review of more than 2,000 studies conducted between 1996 and 2007 found that the average difference in bioequivalence between generic and innovator drugs was 3.5%.
Establishing that the API of a generic drug is bioequivalent to that of the originator drug does not necessarily mean that the medications are exactly the same. For example, a generic tablet may be a different size, shape, or color than the originator product. The U.S. Food and Drug Administration (FDA) also does not require that generic drugs contain the same inactive ingredients (excipients), such as binding materials, flavoring agents, dyes, and preservatives. In effect, it is possible that someone may experience a side effect upon switching from an originator drug to a generic drug, such as an adverse reaction to a particular excipient.
Despite these differences, generics have been confirmed, in various studies, to be therapeutically equivalent to originator drugs. In a Harvard Medical School meta-analysis of 47 clinical trials of cardiovascular drugs, no statistically significant differences in efficacy or safety outcomes were documented among those receiving generic drugs compared with those receiving originator products. A study comparing generic and originator formulations of extended-release clarithromycin for respiratory tract infections also demonstrated similar outcomes. Additionally, comparable clinical outcomes were noted in a large Zambian cohort comparing generic and originator ARVs for HIV infection.
Most ARVs have a relatively wide therapeutic window. If taken correctly, blood concentrations of the drug remain safely above the minimum effective concentration required to be effective and below the minimum toxic concentration required for optimal safety (see figure). In turn, even if a generic ARV’s absorption differs somewhat from that of the originator product, neither efficacy nor safety should be compromised. This is especially true with the standard practice of using regimens containing three or more ARVs to maximize efficacy. And while even a slight upward deviation in a generic ARV’s absorption can potentially increase the risk of serious side effects, this was a much more significant problem with older drugs used to treat HIV (many of which are rarely used in the United States and are being phased out in low- and middle-income countries).
Another key approval requirement for generic drugs undergoing stringent regulatory approval, which includes generic versions of originator drugs to be made available in low-income countries through the President’s Emergency Plan for AIDS Relief through the FDA tentative approval process, are current good manufacturing practices (GMPs). In short, all drug manufacturers must prove that they maintain appropriate facilities, equipment, and staffing, and that they follow strict procedures for producing medicines through every aspect of sterilization, development, testing, production, quality control, and distribution.
GMP enforcement is a major bottleneck for regulatory agencies like the FDA and European Medicines Agency, as they require regular inspections of drug manufacturing facilities. This is a daunting task in light of the fact that the pharmaceutical supply chain has become increasingly globalized and involves numerous API and finished drug manufacturers in various countries, compounded by limited regulatory agency resources and staffing to rapidly and thoroughly conduct the necessary inspections in lockstep with the increasing number of new generic drug approval applications (ANDAs). A consequence of this bottleneck has been a 30-month backlog of the 800 to 900 ANDAs received annually—including those for drugs that have clearly established bioequivalence—which stymies competition among manufacturers required to drive down prices, drains regulatory agency resources, increases costs to generics manufacturers, and decreases patient and provider confidence in the quality of generic products.
In an effort to hasten the delivery of quality-assured generic drugs, the Generic Drug Users Fee Amendments (GDUFA) of 2012 were signed into law by President Obama on July 9, 2012. Comprising a mix of ANDA, backlog, and facility fees paid by API and finished drug manufacturing sites, the legislation provides the FDA with an influx of US$1.5 billion through 2017 to improve the timeliness of generic drug application reviews. GDUFA also aims to enhance the FDA’s ability to protect generic drug users—both domestically and globally—by requiring that U.S. and global manufacturers are held to consistent, high-quality standards and are inspected biennially, with comparable rigor and frequency.
GDUFA’s fees are not, however, without significant concerns. Though they won’t likely hinder manufacturer interest in high-prevalence diseases in the United States, particularly if streamlined FDA approval processes result in expedited revenue returns, the fees are potential barriers when it comes to low-prevalence diseases. Tuberculosis, and to some extent HIV, are prime cases in point. We need to encourage more generic drug manufacturers to seek regulatory approval, not only to ensure multiple sources of essential drugs and to prevent stock-outs, but also to maximize competition and drive down treatment costs. When it comes to low-prevalence diseases, the GDUFA fees forecast by manufacturers may mean even less returns on their investment. For TB programs in the U.S., this would not be a step in the right direction.
The FDA continues to chart its GDUFA implementation plans, including a public hearing that took place on September 17 and a comment period open until October 13. TAG has been actively engaged in these processes, along with several other domestic and global efforts to overcome research, regulatory, and licensing challenges that hinder access to safe, effective, and affordable generic drugs for HIV, HCV, and TB.
