 The discovery of HAART and the push for evidence-based HIV treatment
The discovery of HAART and the push for evidence-based HIV treatment
By Mark Harrington
This is the third in a series looking back at the first two decades of TAG’s work to speed up AIDS research. In Part I: TAG’s early campaigns to reform
the National Institutes of Health (NIH) AIDS research, boost the federal budget, and revitalize HIV basic science research. In Part II: TAG’s response to bad drugs, badly designed clinical trials, and inadequate surrogate markers. Here we look at the rise of highly active antiretroviral therapy (HAART) and the push for evidence-based HIV treatment.
The approval of therapies based on inadequate, ambiguous, uninterpretable or incomplete data offers severe and potentially insurmountable difficulties in the future evaluation of new treatments. This is the deck with which the current therapeutic house of cards was built.
—Spencer Cox, Testimony before the FDA Antiviral Drugs Advisory Committee regarding accelerated approval of stavudine (d4T), May 20, 1994
Prologue
As we pointed out in the previous article in this series about TAG’s accomplishments in its first two decades, the early years of antiretroviral therapy (ART) development recalled Matthew Arnold’s “Dover Beach”: “And we are here as on a darkling plain / Swept with confused alarms of struggle and flight, where ignorant armies clash by night.” The disappointments of the early monotherapy and dual-therapy years between 1987, which saw U.S. Food and Drug Administration (FDA) approval of AZT (Retrovir), and 1995, which saw that of the first protease inhibitor, saquinavir, were succeeded by a difficult-to-believe series of breakthroughs in viral-load monitoring, clinical trials design, and rapid development of new molecules, which came together in 1996 to form the breakthrough known as highly active antiretroviral therapy, or HAART.
The development of HAART, which first came to fruition after fifteen years of basic and ten years of clinical research on HIV treatment, was not rapid, preordained, linear—nor was it always logical. From the contemporary vantage point, with over 32 new chemical entities (NCEs) and fixed-dose combination (FDC) therapies approved by the FDA, it is not always clear what combination of activism, industry, regulation, and research was required to lead to this discovery, which has now resulted in over 8 million people around the world receiving relatively safe, unusually effective combination therapy for a lifelong infectious and almost incurable disease, HIV infection.
Clinical drug development for HIV before HAART can be roughly divided into three periods:
- The first period of HIV research (1981–85) was characterized by its epidemiological identification, the naming (and often, shaming) of risky behaviors and groups, the identification of a causative agent (Paris, 1983; Bethesda, 1984; and San Francisco, 1984), the deployment of an antibody-based blood test to protect the blood supply (1985), and the surprising discovery that an old discarded cancer drug, azidothymidine (AZT), was active in vitro (1984) and in vivo (1985–86) against HIV infection. Most research was academic, carried out at prestigious institutions such as the Insitut Pasteur, where HIV was first discovered, the National Institutes of Health (NIH), and at centers in hard-hit cities such as Los Angeles, New York, and San Francisco.
- The second period of research (1985–90) can be described as the heyday of AZT. First licensed in 1986, it was the only FDA-approved drug for six years—until didanosine’s (ddI’s) approval in mid-1991—and the only “active” HIV drug until the ddI parallel track was launched after national activist pressure in mid-1989. The AZT monopoly price was obscene, at $10,000 a year, the dose was monumentally too high, at 1,500 milligrams/day (250 mg every four hours), causing often-fatal anemia and disabling muscle weakness, and the drug’s effects wore off as resistance emerged after a few weeks to months. The advent of AZT was either a godsend for the new NIH-funded AIDS Clinical Trials Group (ACTG) or a curse, for AZT initially consumed almost 80 percent of the new extramural chain of academic groups’ budget, time, and patients. Some studies (ACTG 001, in people with HIV and Kaposi’s sarcoma) might have been useful, had they been published; others (ACTG 002, comparing two doses) helped—eventually, after pressure from ACT UP—bring down AZT’s dose to 600 mg (300 mg twice daily); and still others (ACTG 016, in “AIDS-related complex” and ACTG 019, in earlier HIV infection), stopped early, led to the premature and later discredited (Anglo-French Concorde study, 1993) recommendation that all those with HIV and CD4 counts below 500 cells/mm3 commence therapy.Thousands probably took the drug too early, developing resistance; thousands of others suffered unnecessary toxicities; and some probably benefited from AZT long enough to obtain other therapies. But it was a very mixed bag. And for the next few years, most of the clinical trials action seemed to be on AZT-like drugs such as ddI (FDA approved in 1991), zalcitabine (ddC; 1992), stavudine (d4T; 1994), and lamivudine (3TC; 1995). All but AZT were approved based on small and short-term changes in parameters such as CD4 counts (ddI was notoriously approved based on a small increase of just 10 CD4 cells that lasted for a matter of weeks), weight gain, and improved Karnofsky performance score. But the obvious profitability of AZT soon induced other companies to enter the drug discovery area, and some began exploring targets different from AZT.
Finally, this period saw the almost complete exclusion of research on the AIDS-related infections and cancers that were actually the proximate cause of death for most people with AIDS—conditions such as Pneumocystis carinii pneumonia (PCP), cytomegalovirus (CMV) retinitis and colitis, cerebral toxoplasmosis, cryptococcal meningitis, disseminated candidiasis, Kaposi’s sarcoma (KS), B-cell lymphomas, and so on. There were many potential treatments and even preventive agents for these conditions, but the NIH-funded efforts ignored most of them. Community-based groups sprung up in San Francisco, New York, and elsewhere to study some of these preventive and therapeutic approaches to opportunistic infections (OIs) and also to study agents that did not have clearly demonstrated and biologically plausible anti-HIV mechanisms of activity, such as AL-721, Ampligen, dextran sulfate, isoprinosine, ribavirin, or the notorious trichosanthin (Chinese cucumber root, a.k.a. “Compound Q”).
There was also an interest in treating AIDS with immunologically active agents such as alpha interferon, gamma interferon, interleukin-2 (IL-2), and some more-or-less quack approaches like compounds such as “Imreg-1″ and “Imreg-2″ (I will never forget Spencer Cox sitting at a table at the East Village diner Odessa—it could have been Leshko’s—asking Imreg-1’s “discoverer” and manufacturer, “What’s actually in the drug, Dr. Gottlieb?” and our dismay when the response was, “We don’t actually know; it’s a made from a leukocyte cell lysate [i.e., by grinding up blood cells and extracting something somehow].”
- The third period of HIV treatment research (1990–95) followed directly upon the ddI parallel track and ACT UP’s “Storm the NIH” demo, which led not only to the full representation of activists and people with HIV on all the ACTG research committees, but to much accelerated and expanded research to prevent and treat AIDS-related opportunistic infections and cancers, such that by the mid-1990s many of them could be effectively prevented (PCP, disseminated fungal disease, toxoplasmosis), or at least treated in some cases (cutaneous KS, some B-cell lymphomas, CMV). The ACTG tooled around endlessly with various one- and two-drug combinations, usually nucleoside reverse transcriptase inhibitors only, sometimes adding in another class, often because its sponsors (e.g., Roche and Boehringer Ingelheim) lacked the guts or resources to self-develop the drug. The nadir of the period was the debacle of ACTG 155 at Berlin in mid-1993. By the end of 1995, however, some combination studies (ACTG 175) demonstrated clinical benefit, while other new agents—ritonavir from Abbott, indinavir from Merck, and nelfinavir from Agouron—began to show unexpected potency when measured with new quantitative viral-load assays.
The Beginning of the Modern Era
Soon after AZT’s approval by the FDA in March 1987, it became clear that its benefits were transient and limited by severe anemia and other toxicities, and that treatment failure was associated with the emergence of HIV resistant to the drug. Thus, even in 1989, it seemed obvious to Burroughs Wellcome’s leading virologist, David Barry, who led the AZT development team, that “you’re going to have four or five drugs for the OIs and two, three and maybe four drugs for antivirals.”
Dr. Barry’s prediction was right, and a number of companies began fortifying their pipelines. Manufacturers either bought potential HIV agents, as did Bristol-Myers with ddI and Hoffmann-La Roche with ddC; licensed them, as Bristol did with d4T; or set out to modify existing renin inhibitors (a class of blood pressure medications industry was attempting to develop from the 1970s on) to develop aspartic protease inhibitors (PIs), that could bind and inhibit the protease enzyme of HIV-1. Researchers from a number of companies including Abbott, Merck, and Roche began efforts to crystallize the HIV-1 protease enzyme and to screen compounds that blocked its activity, as did the National Cancer Institute (NCI).
Early in 1990, I remember gazing in wonder at a giant model of the crystallized protease enzyme published by the NCI in 1989, with little sense of whether its therapeutic promise as a target was likelier to become science or science fiction. The first PI wouldn’t be approved until late 1995. We’ll never know whether a sensible, directed research approach—if that isn’t itself an oxymoron—could have accelerated this discovery.
By summer 1994, TAG and allies such as David Barr and Derek Link at GMHC and activist Carlton Hogan from the University of Minnesota clinical trials coordinating center had come to believe that a new approach to AIDS clinical trials was necessary, one that allowed flexibility in the control arm (participants could take whatever approved or parallel-track HIV drug they wanted) but that restored rigor to the process by randomizing participants to receive a new PI or a placebo at a 2:1 ratio—and that used clinical endpoints, notably time to an AIDS-defining event or death—rather than relying on the discredited surrogate marker of CD4 cell-count changes due to therapy.
We then released our report, Rescuing Accelerated Approval: Moving Beyond the Status Quo, which was distributed at a contentious FDA advisory committee hearing in September 1994 (reviewed in “On a Darkling Plain” in the October 2012 TAGline).
TAG and our allies worked assiduously to watchdog the phase II/III clinical trials of every company that was making an HIV PI, including Abbott’s ritonavir, Agouron’s nelfinavir, Merck’s indinavir, and Roche’s saquinavir. TAG’s recommendations to regulators and companies were published in the February 1995 report Problems with Protease Inhibitor Development Plans.
In summer 1995, Spencer Cox published his scathing, still-compelling report FDA Regulation of Anti-HIV Drugs: A Historical Perspective—a cautionary retrospective on the first, mostly unsuccessful, ten years of HIV drug development and regulation.
Drug Combinations to the Fore
In September 1995, Spencer Cox, Michael Marco, Tim Horn, and I attended the 35th ICAAC, where the results of AIDS Clinical Trials Group study 175 were presented, along with early phase I viral-load data from Abbott’s ritonavir development program.
ACTG 175 was the first study to prove, using clinical endpoints, that ddI alone, AZT plus ddI, or AZT plus ddC were better than AZT alone, in both AZT-naive and -experienced persons, in terms of slowing progression to AIDS or death.
“I mean, it’s not like I live for bad news. It looks like we’re making some progress,” commented Spencer Cox in TAG Does ICAAC.
An early Abbott combination study of ritonavir, AZT, and ddC appeared to show even more arresting data: According to lead author Daniel Norbeck, the regimen yielded a CD4 count increase of 110 cells and an unprecedented 2.5 log decrease in viral RNA lasting for the 20 weeks of the study. Over the subsequent weeks, Norbeck claimed, an increasing proportion of participants became viral culture–negative—which is to say they could not culture infected cells from the blood.
The era of monotherapy was on its way out.
In November 1995, the FDA advisory committee met to review Roche’s application for accelerated approval of saquinavir, Glaxo Wellcome’s for accelerated approval of lamivudine (3TC), and Bristol-Myers’s for full approval of stavudine (d4T). The three-day hearing was neither pleasant nor terribly informative. Moving to the combination-therapy era without the proper monitoring tools—e.g., quantitative viral-load testing, which came of age only the following year—made it difficult to assess the benefit of the two AZT-like drugs and that of the first PI, saquinavir.
Nonetheless, we believed that Roche had met our demands from the previous year, notably: adequate and well-controlled clinical endpoints studies were under way to show whether the drug could prolong disease-free time or survival; the completion or implementation of studies to demonstrate a favorable combination of changes in CD4 levels and viral load; evidence adequately characterized and acceptable safety evidence; and efforts to enroll an expanded access program.
Therefore, we supported approval of saquinavir, despite its low dose and the lack of definitive clinical endpoint data. The drug was approved in December 1995. Less than two years later, Roche admitted that the licensed dose was suboptimal, though it didn’t take responsibility for exposing people to a greater risk of cross-resistance to other, more potent PIs such as ritonavir or indinavir. Ultimately, Roche developed a more bioavailable dose that could have competed with the stronger PIs, but it was too late; the drug was doomed by the company’s early mistakes.
Luckily, the FDA had taken proactive steps to expedite its review of the next two PIs in the pipeline, both of them more potent than saquinavir: Abbott’s ritonavir and Merck’s indinavir.
Abbott had adopted Spencer Cox’s recommendation (from the 1994 Rescuing Accelerated Approval: Moving Beyond the Status Quo report):
After discussing the proposed expanded access program “for people who have failed or proven intolerant to all available AIDS drugs, or who have under 50 T-cells, TAG proposes a standard expanded access program, in which all patients would receive protease inhibitor, but would be randomly assigned to either high-dose or low-dose….
For other people with HIV, TAG has proposed a “large, simple trial.” Essentially, all HIV-positive people would be eligible for participation, with those above and below 200 T-cells studied separately. Study participants would be randomly assigned to take either one of two protease inhibitors or placebo, or one of two different protease products and placebo. Other than their study treatment, participants would be able to take any other drug they wanted, approved or unapproved, and to pursue the best medical care.
Annus Mirabilis
On February 1, at the 3rd Conference on Retroviruses and Opportunistic Infections in Washington, D.C., Abbott showed the results of the ritonavir study. In just six months, those receiving ritonavir plus standard of care (SOC) had 50% fewer deaths than those receiving placebo plus SOC. Spencer, attending CROI, was in tears: “We’re going to live!” (see figure).
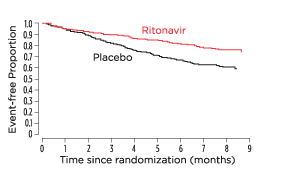
Evidence of survival with a protease inhibitor. A Kaplan-Meier analysis demonstrating the proportion of subjects who survived and remained free of a new AIDS-defining diagnosis while being treated with either ritonavir or placebo. Exclaimed TAG’s first Antivirals Project Director upon seeing these results for the first time in 1996: “We’re going to live!” Adapted from Cameron et al. Lancet. 1998 Feb 21;351(9102):543–9.
On February 29, 1996, the FDA advisory committee reviewed ritonavir. Spencer Cox was the ad hoc community representative on the panel. The committee resoundingly recommended approval. On March 1, 1996, the FDA approved ritonavir. Merck’s indinavir was approved two weeks later, Roche’s Amplicor-brand RT-PCR test for HIV RNA was approved three months later, and on June 21, the FDA granted accelerated approval to the first-ever non-nucleoside reverse transcriptase inhibitor, Boehringer Ingelheim’s nevirapine (Viramune).
The media became a bit giddy. At the end of June, theEconomist cover story read: “A Solution for AIDS?”
Everything culminated at the 11th International AIDS Conference in Vancouver, British Columbia, in early July 1996. John Mellors presented his famous paper from the Multicenter AIDS Cohort Study (MACS) showing that viral load predicted the rate of CD4 decline. Virologist John Coffin delivered his famous metaphor comparing the HIV-infected patient to a train speeding along a track towards a deadly cliff—the viral load conveying the speed of the train, with CD4 count measuring the distance to the cliff.
On July 11, 1996, the last day’s late-breaker presentations confirmed the overwhelming and unexpected benefit of triple-combination therapy when initiated simultaneously in people who had never received the drugs in question. Six researchers from four teams showed results from six studies of five different regimens that reduced HIV RNA levels to undetectable levels in the plasma.
One of the most dramatic presentations was made by David Ho of the Aaron Diamond AIDS Research Center (ADARC), presenting on 12 antiretroviral-naive individuals receiving AZT, 3TC, and nelfinavir. CD4 counts started at 245 and viral load at 56,000. CD4 counts rose by about 100 cells, while “at twelve weeks, all eleven patients remaining on the study had levels below that threshold [25 HIV RNA copies/ml], and predicted that they were essentially at zero.”
As I wrote in Viral Load in Vancouver:
The room became very quiet. You could have heard a pin drop. A collective silent sigh ensued, as the full implications of this sunk in to the thousands of scientists, reporters and activists assembled on this last late-breaker session of the Vancouver conference. People I knew and loved were in this study. Their viral load had been reduced, within three months, to virtually zero. Perhaps some of us would live, after all.
On August 6, I went down to the NIH Clinical Center for my second lymph-node biopsy. My viral load was 76,790 (Chiron bDNA). The next day, I started my first antiretroviral treatment: 3TC, d4T, and indinavir. By August 23, my viral load had dropped to 2,932 (PCR). By December, my CD4 cells had risen from 152 to 597, and my viral load was undetectable.
Not everyone fared as well. Spencer Cox, who was extensively antiretroviral-experienced, mostly with nucleoside analogues, initially responded well to ritonavir but then developed resistance. In midsummer, he switched to indinavir, yet by late September his viral load was virtually back to baseline, at 400,000, just seven months after starting ritonavir. We all feared this trajectory was a harbinger of dangers that might be ahead for everyone who appeared to benefit in the short-term from triple-combination therapy.
On September 21, Tae-Wook Chun, then of Robert Siliciano’s lab at Johns Hopkins, later at NIAID, lectured at ADARC on cellular latency of integrated HIV provirus in resting CD4 cells. This prefigured the end of the eradication theory put forth by David Ho and Alan Perelson earlier in the year, by which triple therapy given for a few years might cure HIV.
Two days later, we met Mike Saag from the University of Alabama at Birmingham to talk about his proposed START protocol (“Strategic Timing of ART,” ACTG 355). This study would be labeled “overly ambitious” by the ACTG and withdrawn in March 1997. The ACTG would never do a “when to start” study, and 17 years later we still don’t know the best time to initiate ART for individual benefit.
That fall, the Department of Health and Human Services convened a Public Health Service panel to develop clinical-practice guidelines for HIV. Both Spencer Cox and I were named to the panel, which was to develop the first guidelines for the use of highly active antiretroviral therapy (HAART).
Laurie Garrett’s cover story in Newsday, “The Curse of the Cure,” rounded out the year on December 17. Spencer and I were on the cover. Which one had the drug-resistant HIV? And how long would the benefits of HAART last for anyone, drug-experienced or drug-naive?
At the end of that year, I wrote in TAGline:
On a public health level, assuring access to and information about new treatment strategies is an enormous task even in the developed world, and the chances of extending their use to the developing world where over 90% of HIV cases occur appear slender until and unless a new global commitment to providing health care to all emerges. Given the political landscape in the USA, where even its own citizens are routinely denied access to health care, this prospect seems remote. More likely is the recapitulation with HIV of what occurred with tuberculosis, when several generations of effective drugs were wasted by inadequate public health efforts, resistance to all of them emerged, and, after a hiatus of several decades, tuberculosis returned with a vengeance in a new, multi-drug resistant form, its re-emergence amplified by the widespread immune dysfunction triggered by the HIV pandemic.
